Overview
We investigate the molecular mechanisms underlying frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS). We are particularly interested in the C9orf72 gene, which is a common cause of both FTD and ALS and the CHMP2B gene, which is a rare cause of FTD. We use a multidisciplinary approach including disease modelling in mice, Drosophila, primary neurons and iPS cell-derived neurons. We combine these model systems with analysis of patient tissue in order to ensure the relevance of our work to the disease process we are studying. We are also working to translate our basic research into potential therapies and are currently developing small molecule and gene therapy therapeutics based on our findings. We collaborate with both academic and pharmaceutical partners in order to achieve these goals.
C9orf72
Background
A GGGGCC repeat expansion in C9orf72 is the most common known cause of FTD and ALS. Three potential mechanisms have been proposed. 1) Loss of function of C9orf72. 2) Toxicity of repeat RNA through sequestration of RNA binding proteins. 3) Toxicity of dipeptide repeat proteins generated by repeat-associated non-ATG (RAN) translation.
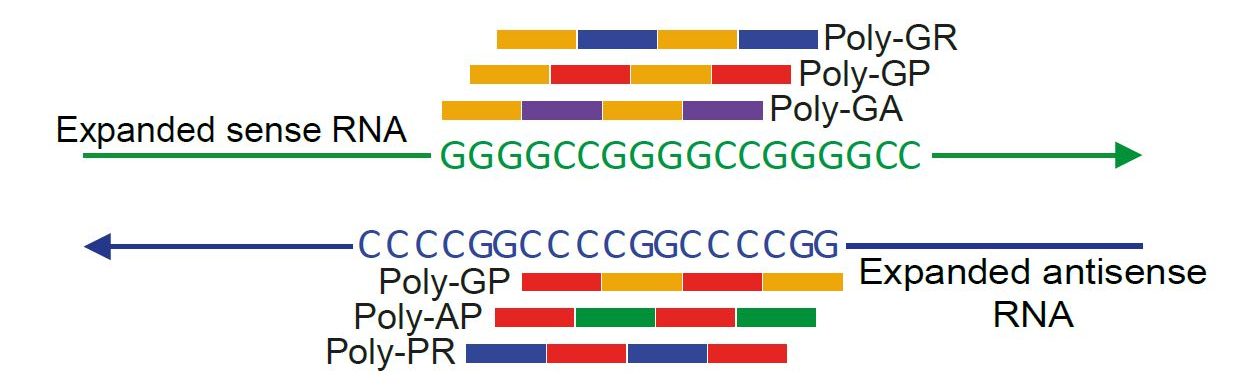
RAN translation of C9orf72 GGGGCC repeats. RAN translation occurs in all three frames in both the sense and antisense directions, leading to the generation of proteins composed of repeating dipeptides: poly(GR), poly(GA) and poly(GP) in the sense direction and poly(AP), poly(PR) and poly(GP) in the antisense direction. Note poly(GP) is made in both the sense and antisense direction. G- glycine, R – arginine, A – alanine, P – proline.
Our work on C9orf72 broadly falls into two themes i) investigating mechanisms of toxicity and ii) developing novel therapeutic approaches.
Mechanisms of toxicity
We showed, in collaboration with Linda Partridge’s group at UCL, that the dipeptide repeat proteins poly(GR) and poly(PR) are profoundly neurotoxic in Drosophila (Mizielinska et al Science 2014). We also showed that aggregates of RNA (termed RNA foci) derived from both sense and antisense GGGGCC repeat RNA are frequently observed in frontal cortex neurons in people with the C9orf72 repeat expansion (Mizielinska et al 2013). We are now using a range of approaches to further investigate both protein and RNA toxicity. In our Drosophila models these include large-scale modifier screens, mass spectrometry and transcriptomics. We are coupling these analyses with investigation of induced pluripotent stem cell (iPSC)-derived neurons that carry the C9orf72 repeat expansion, with a particular focus on high-content imaging. We are also utilising bioinformatics approaches, in collaboration with Vincent Plagnol and Pietro Fratta at UCL, to further probe disease mechanisms. These complementary approaches may identify the key regulators and mediators of C9orf72 dipeptide repeat-induced neurodegeneration.
iPSC-motor neurons
Therapeutic approaches
We showed that the C9orf72 GGGGCC repeat RNA forms a secondary structure termed a G-quadruplex (Fratta et al 2012). We worked with the UCL School of Pharmacy to develop small molecules which specifically bind the GGGGCC repeat RNA G-quadruplex. These small molecules reduced both repeat-derived RNA foci and RAN translation (Simone, Balendra et al 2018).
We are now building on this work to develop new gene therapy and small molecule approaches to target mechanisms implicated in C9orf72-FTD/ALS as well as neurodegeneration more broadly. We collaborate with the Alzheimer’s Research UK UCL Drug Discovery Institute to achieve these aims.
CHMP2B
A mutation in the CHMP2B gene causes frontotemporal dementia (FTD) in a large Danish family. CHMP2B is a member of the endosomal sorting complex required for transport-III (ESCRT-III). ESCRT proteins were first shown to be required for delivering endocytosed proteins to lysosomes for degradation. This is achieved through mediating the formation within endosomes of intraluminal vesicles that contain the cargoes to be degraded. On fusion with lysosomes the proteins within the intraluminal vesicles are degraded. ESCRTs are also required for effective autophagy, potentially through cross-talk with the endo-lysosomal pathway. Remarkably, ESCRT-III proteins are also utilised for other membrane scission events with similar topology to intraluminal vesicle formation, such as virus release, cytokinesis, nuclear envelope reformation and potentially autophagosome closure.
We have shown that mutant CHMP2B impairs both the endo-lysosomal and autophagy pathways (Filimonenko et al 2007; Urwin et al 2011) and that mutant CHMP2B rather than loss of the normal function of CHMP2B is likely to drive the disease process (Ghazi-Noori et al 2013). More recently we have identified lysosomal aggregates similar to those observed in lysosomal storage diseases as the main neuronal pathology in CHMP2B-FTD (Clayton et al 2015). Interestingly, other major FTD genes such as progranulin and TMEM106B also have important roles in lysosome function. Therefore lysosomal dysfunction may be a central causative pathway in FTD. A key unanswered question is how lysosomal dysfunction specifically affects neurons. We are therefore studying the effects of mutant CHMP2B and related FTD genes in cortical neurons. We are using a range of techniques to study both neuronal and lysosomal functions including live imaging of endo-lysosomes in primary and iPSC-derived cortical neurons. We recently found that knockdown of TMEM106B can ameliorate some of the deleterious effects of mutant CHMP2B in neurons (Clayton et al 2018).
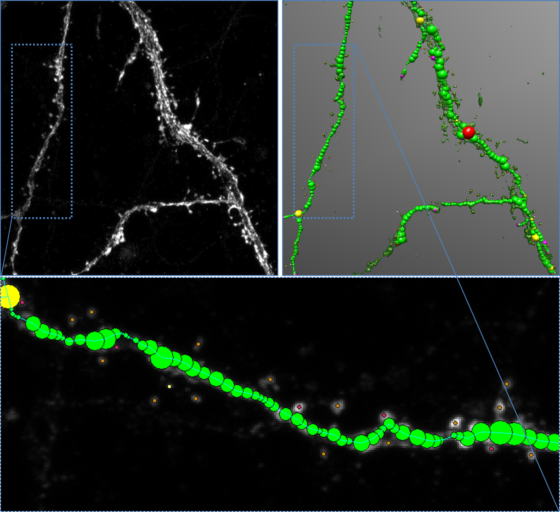
Automated analysis of dendritic spines in cultured neurons
References
Clayton EL, Mizielinska S, Edgar JR, Nielsen TT, Marshall S, Norona FE, Robbins M, Damirji H, Holm IE, Johannsen P, Nielsen JE, Asante EA, Collinge J; FReJA consortium, Isaacs AM. Frontotemporal dementia caused by CHMP2B mutation is characterised by neuronal lysosomal storage pathology. Acta Neuropathol. 2015 Oct;130(4):511-23.
Clayton EL, Milioto C, Muralidharan B, Norona FE, Edgar JR, Soriano A, Jafar-Nejad P, Rigo F, Collinge J, Isaacs AM. Frontotemporal dementia causative CHMP2B impairs neuronal endolysosomal traffic-rescue by TMEM106B knockdown. Brain. 2018 Dec 1;141(12):3428-3442.
Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L, Fisher EM, Isaacs A, Brech A, Stenmark H, Simonsen A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol. 2007; 179(3):485-500.
Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EMC, Parkinson GN, Isaacs AM. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep. 2012; 2:1016.
Ghazi-Noori S, Froud KE, Mizielinska S, Powell C, Smidak M, Fernandez de Marco M, O’Malley C, Farmer M, Parkinson N, Fisher EM, Asante EA, Brandner S, Collinge J, Isaacs AM. Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. Brain. 2012; 135(3):819-32.
Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IO, Pietrzyk J, Cleverley K, Nicoll AJ, Pickering-Brown S, Dols J, Cabecinha M, Hendrich O, Fratta P, Fisher EM, Partridge L, Isaacs AM. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014; 345:1192-1194.
Mizielinska S, Lashley T, Norona F, Clayton EL, Ridler CE, Fratta P, Isaacs AM. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013; 126(6):845-57.
Simone R, Balendra R, Moens TG, Preza E, Wilson KM, Heslegrave A, Woodling NS, Niccoli T, Gilbert-Jaramillo J, Abdelkarim S, Clayton EL, Clarke M, Konrad MT, Nicoll AJ, Mitchell JS, Calvo A, Chio A, Houlden H, Polke JM, Ismail MA, Stephens CE, Vo T, Farahat AA, Wilson WD, Boykin DW, Zetterberg H, Partridge L, Wray S, Parkinson G, Neidle S, Patani R, Fratta P, Isaacs AM. G-quadruplex-binding small molecules ameliorate C9orf72 FTD/ALS pathology in vitro and in vivo. EMBO Mol Med. 2018 Jan;10(1):22-31.
Urwin H, Authier A, Nielsen JE, Metcalf D, Powell C, Malcolm DS, Holm IE, Johannsen P, Brown JM, Fisher EMC, van der Zee J, the FReJA Consortium, Van Broeckhoven C, Collinge J, Brandner S, Futter C, Isaacs AM. Disruption of Endocytic Trafficking in Frontotemporal Dementia with CHMP2B Mutations. Hum Mol Genet. 2010; 19(11):2228-38.