Overview
We investigate the molecular mechanisms that cause frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) also known as motor neuron disease. We are harnessing this knowledge to develop new treatments. We are particularly interested in the C9orf72 gene, which is a common cause of both FTD and ALS and the CHMP2B gene, which is a rare cause of FTD. We use a multidisciplinary approach including disease modelling in mice, Drosophila and induced pluripotent stem cell (iPSC)-derived neurons, which we combine with analysis of biosamples from people with FTD and ALS. We are working to translate our basic research into potential therapies and are currently developing small molecule and gene therapy approaches for FTD and ALS. In addition, we develop biomarkers to enable clinical trials of new treatments.
C9orf72
Background
A GGGGCC repeat expansion in C9orf72 is the most common known cause of FTD and ALS. Three potential mechanisms have been proposed. 1) Loss of function of C9orf72. 2) Toxicity of repeat RNA through sequestration of RNA binding proteins. 3) Toxicity of dipeptide repeat proteins generated by repeat-associated non-ATG (RAN) translation.
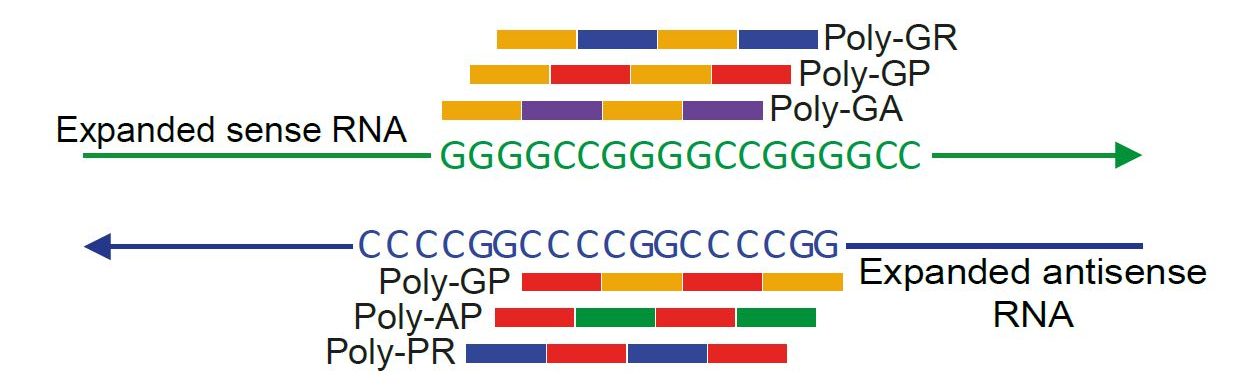
RAN translation of C9orf72 GGGGCC repeats. RAN translation occurs in all three frames in both the sense and antisense directions, leading to the generation of proteins composed of repeating dipeptides: poly(GR), poly(GA) and poly(GP) in the sense direction and poly(AP), poly(PR) and poly(GP) in the antisense direction. Note poly(GP) is made in both the sense and antisense direction. G- glycine, R – arginine, A – alanine, P – proline.
Research
We showed, in collaboration with Linda Partridge’s group at UCL, that the dipeptide repeat proteins poly(GR) and poly(PR) are profoundly neurotoxic in Drosophila (Mizielinska et al Science 2014). We also showed that aggregates of RNA (termed RNA foci) derived from both sense and antisense GGGGCC repeat RNA are frequently observed in frontal cortex neurons in people with the C9orf72 repeat expansion (Mizielinska et al 2013). We are now using a range of approaches to further investigate both protein and RNA toxicity. In our Drosophila models these include large-scale modifier screens, transcriptomics and lipidomics (e.g. Moens et al 2019; Atilano et al 2021; Giblin et al 2024). We perform extensive research in iPSC-neurons (e.g. Simone et al 2018; Moens et al 2019; Milioto et al 2024; Giblin et al 2024) including validation of findings from our Drosophila and mouse models, lipidomics and the use of genome editing techniques to build reporter lines for high-throughput screening.
We are developing AAV-based gene therapy approaches with academic and industry partners to either reduce sense and antisense repeat RNAs (Kempthorne et al 2024) or provide neuroprotection from the damaging dipeptide repeat proteins.
We also work closely with the Alzheimer’s Research UK UCL Drug Discovery Institute to perform high-throughput screening and medicinal chemistry optimisation to develop small molecules to treat C9orf72 FTD/ALS and sporadic FTD/ALS.
To support our therapy development for C9orf72 FTD/ALS we also developed an ultra-sensitive Simoa assay to detect polyGP in patient CSF (Wilson et al 2022) that was used in Wave Life Sciences’ FOCUS-C9 clinical trial.
iPSC-motor neurons
CHMP2B
A mutation in the CHMP2B gene causes frontotemporal dementia (FTD) in a large Danish family. We have studied FTD-CHMP2B for many years and co-lead an international collaboration between the UK, Denmark and Sweden that has published >20 papers on the clinical and molecular aspects of the disease (e.g. Clayton et al 2015). CHMP2B is a member of the endosomal sorting complex required for transport-III (ESCRT-III). ESCRT proteins form a membrane remodelling complex required for many cellular processes including endosome intraluminal vesicle formation, cytokinesis, nuclear envelope reformation and lysosome and plasma membrane repair. We are currently developing gene therapy approaches to treat CHMP2B-FTD in collaboration with the Nucleic Acid Therapy Accelerator. We also have an exciting long-term collaboration Jez Carlton’s lab at King’s College London to investigate the role of FTD/ALS genes, including CHMP2B, on lysosome and plasma membrane repair. With funding from the Chan Zuckerberg Initiative, we are performing whole-genome CRISPR screening in iPSC-neurons to discover new components of the lysosome and plasma membrane repair machinery in neurons.
References
Atilano ML, Grönke S, Niccoli T, Kempthorne L, Hahn O, Morón-Oset J, Hendrich O, Dyson M, Adams ML, Hull A, Salcher-Konrad MT, Monaghan A, Bictash M, Glaria I, Isaacs AM*, Partridge L*. Enhanced insulin signalling ameliorates C9orf72 hexanucleotide repeat expansion toxicity in Drosophila. Elife. 2021 Mar 19;10:e58565. doi: 10.7554/eLife.58565. PMID: 33739284.
Clayton EL, Mizielinska S, Edgar JR, Nielsen TT, Marshall S, Norona FE, Robbins M, Damirji H, Holm IE, Johannsen P, Nielsen JE, Asante EA, Collinge J, None None & Isaacs AM. Frontotemporal dementia caused by CHMP2B mutation is characterised by neuronal lysosomal storage pathology. Acta Neuropathol. 2015 Sep;130(4):511-23 doi: 10.1007/s00401-015-1475-3. PMID: 26358247.
Giblin A, Cammack AJ, Blomberg N, Mikheenko A, Carcole M, Coneys R, Zhou Z, Mohammed Y, Olivier D, Atilano M, Niccoli T, Coyne A, van der Kant R, Lashley T, Giera M, Partridge L, Isaacs AM. Neuronal polyunsaturated fatty acids are protective in ALS/FTD. bioRxiv 2024.01.16.575677; doi: https://doi.org/10.1101/2024.01.16.575677.
Milioto C, Carcolé M, Giblin A, Coneys R, Attrebi O, Ahmed M, Harris SS, Lee BI, Yang M, Ellingford RA, Nirujogi RS, Biggs D, Salomonsson S, Zanovello M, de Oliveira P, Katona E, Glaria I, Mikheenko A, Geary B, Udine E, Vaizoglu D, Anoar S, Jotangiya K, Crowley G, Smeeth DM, Adams ML, Niccoli T, Rademakers R, van Blitterswijk M, Devoy A, Hong S, Partridge L, Coyne AN, Fratta P, Alessi DR, Davies B, Busche MA, Greensmith L, Fisher EMC, Isaacs AM. PolyGR and polyPR knock-in mice reveal a conserved neuroprotective extracellular matrix signature in C9orf72 ALS/FTD neurons. Nat Neurosci. 2024 Apr;27(4):643-655. doi: 10.1038/s41593-024-01589-4. PMID: 38424324.
Kempthorne L, Vaizoglu D, Cammack AJ, Carcolé M, Suklai P, Muralidharan B, Kroll F, Moens TG, Yshii L, Verschoren S, Hölbling BV, Katona E, Mikheenko A, Coneys R, de Oliveira P, Zhang Y, Jansen K, Daughrity LM, McGown A, Ramesh TM, Van Den Bosch L, Rahim AA, Petrucelli L, Rihel J, Isaacs AM. Dual-targeting CRISPR-CasRx reduces C9orf72 ALS/FTD sense and antisense repeat RNAs in vitro and in vivo. bioRxiv 2024.01.26.577366; doi: https://doi.org/10.1101/2024.01.26.577366.
Moens TG, Niccoli T, Wilson KM, Atilano ML, Birsa N, Gittings LM, Holbling BV, Dyson MC, Thoeng A, Neeves J, Glaria I, Yu L, Bussmann J, Storkebaum E, Pardo M, Choudhary JS, Fratta P, Partridge L & Isaacs AM. C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathol. 2019 Jan;137(3):487-500 doi: 10.1007/s00401-018-1946-4. PMID: 30604225.
Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P & Isaacs AM. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013 Oct;126(6):845-57 doi: 10.1007/s00401-013-1200-z. PMID: 24170096.
Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IOC, Pietrzyk J, Cleverley K, Nicoll AJ, Pickering-Brown S, Dols J, Cabecinha M, Hendrich O, Fratta P, Fisher EMC, Partridge L & Isaacs AM. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014 Aug;345(6201):1192-1194 doi: 10.1126/science.1256800. PMID: 25103406.
Simone R, Balendra R, Moens TG, Preza E, Wilson KM, Heslegrave A, Woodling NS, Niccoli T, Gilbert-Jaramillo J, Abdelkarim S, Clayton EL, Clarke M, Konrad MT, Nicoll AJ, Mitchell JS, Calvo A, Chio A, Houlden H, Polke JM, Ismail MA, Stephens CE, Vo T, Farahat AA, Wilson WD, Boykin DW, Zetterberg H, Partridge L, Wray S, Parkinson G, Neidle S, Patani R, Fratta P & Isaacs AM. G-quadruplex-binding small molecules ameliorate C9orf72 pathology in vitro and in vivo. EMBO Mol Med. 2018 Jan;10(1):22-31. doi: 10.15252/emmm.201707850. PMID: 29113975.
Wilson KM, Katona E, Glaria I, Swift IJ, Sogorb-Esteve A, Heller C, Bouzigues A, Heslegrave AJ, Patil S, Mohapatra S, Liu Y, Goyal J, Sanchez-Valle R, Laforce R, Synofzik M, Rowe JB, Finger E, Vandenberghe R, Butler CR, Gerhard A, van Swieten J, Seelaar H, Borroni B, Galimberti D, de Mendonça A, Masellis M, Tartaglia C, Otto M, Graff C, Ducharme S, Malaspina A, Zetterberg H, Boyanapalli R, Rohrer JD, Isaacs AM. Development of a sensitive trial-ready poly(GP) CSF biomarker assay for C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2022 Jul;93(7):761-771. doi: 10.1136/jnnp-2021-328710. PMID: 35379698.
For a full list of publications see our publications page